Quality control is a critical step to every workflow. For WGS, this begins at the intake of samples and follows through the entire experiment. Here we will briefly review input QC and post-sequencing QC.
Input QC
Input QC is measure by both quality and quantity of the DNA.
Quality
Quality of input DNA is often a product of the DNA extraction protocol. This can be measured in a few facets. First is use of gel electrophoresis. This will allow you to view the quality in terms of fragmentation. For a short read WGS experiment, this is not as critical as you will be fragmenting the DNA anyways. More important is a measurement that can capture potential contaminants in the DNA prep that may inhibit library generation. Primarily you could utilize a Nanodrop or similar optically based tool to read the optical density. Using metrics like 260/280 and 230/260 ratios can help view possible protein or other contaminants.
In most cases, a 260/280 ratio 1.8-2.0 is considered high quality DNA.
Quantity
Quantity is naturally important, if you dont have enough material you wont be able to generate effective libraries, or you would need to perform an excessive amount of PCR. Quantity can be measured using a Nanodrop as well, though this can often be misleading. A more accurate measurement would be something fluorescence binding based like Qubit. This would enable quantification of just the dsDNA.
For most library preps, 500ng would be a suitable target for submitting to a genomics service provider. Some kits can go to ~1ng as input, and others would require more than 1.5ug.
FASTQC
Quality of the sequencer reads is just as important. This informs much of the strategy going forward into analysis. One primary tool for checking FASTQ QC is called FASTQC. A few of the metrics to check are below- but this is by no means a comprehensive list.
FASTQ quality
FASTQ quality is measured in terms of accuracy per base call, usually presented in Q30 (or Q##). A base with a score of >Q30 means the accuracy of that base call is >99.9%, and this can be viewed on per base or average for a read.
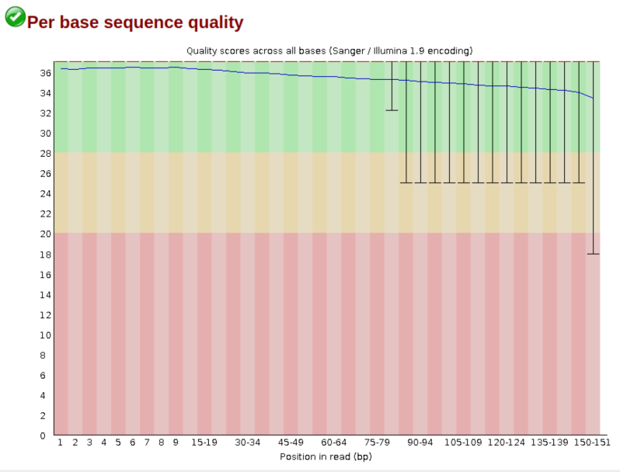
One relic of sequencing is that as the reads continue the quality will drop. This is based on the loss of fidelity of the polymerase and base incorporation over time. This is usually not a major concern in a standard 300 cycle kit, but in the longer 600 cycle kits you can start to see extreme quality drops in the later parts of read 2.
FASTQ GC content
GC content can be used to check both the quality of the fragmentation and the appropriate ratio to your organism. If you are trying to sequence an A-T rich species and you see a high G-C ratio here- something may have been mixed up. You also expect to see a fairly even distribution if the fragmentation was truly random.

Adapter contamination
If your insert is smaller than your sequencing cycle count, you could read through the entire insert and into the adapter regions. This step helps to identify the common regions of adapters to inform you to trim those regions prior to analysis. This example is what a good output looks like that had no adapter contamination.